 by
by Idiopathic Pulmonary Fibrosis (IPF) is a progressive lung disease characterized by scarring (fibrosis) of the lungs for unknown reasons. As the lungs become scarred, it becomes difficult to breathe and transfer oxygen to the bloodstream. While its cause remains unknown, IPF is not contagious or hereditary. In this article, we discuss what IPF is, its symptoms, risk factors, diagnosis and available treatment options.
What is IPF?
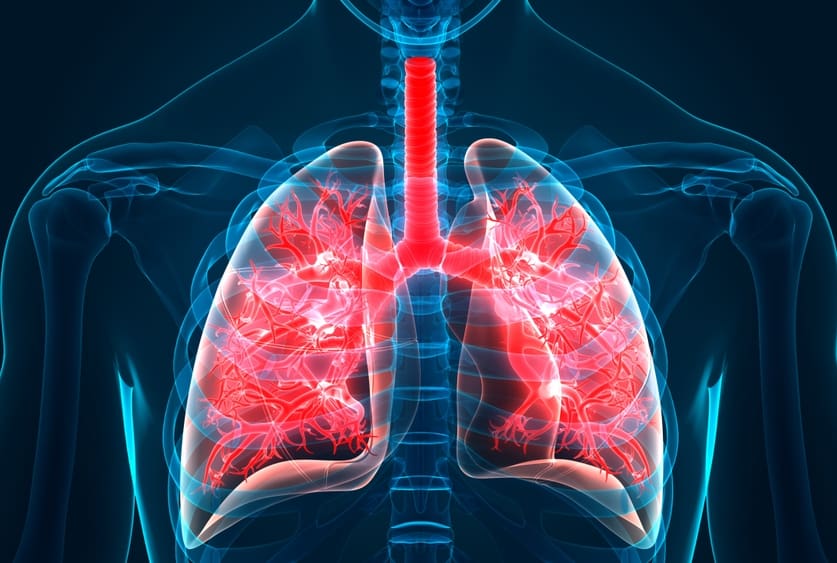
Idiopathic Pulmonary Fibrosis, also referred to as IPF, is a fibrosing interstitial pneumonia of unknown cause that results in progressive scarring of the lungs. Over time, lung tissue is replaced by scar tissue which makes breathing difficult for those affected. As the scarring worsens over months or years, lung function gradually deteriorates leading to respiratory failure. IPF predominantly affects individuals over the age of 50 and is slightly more common in men than women. Its exact prevalence is unknown but it is estimated that over 200,000 individuals globally are affected by IPF.
Symptoms of IPF
Some of the common symptoms experienced in the early stages of IPF include breathlessness or dyspnea during physical exertion, non-productive cough and fatigue. As the condition progresses, symptoms like worsening shortness of breath, weaker cough, finger clubbing, dry mouth and weight loss may occur. Symptoms tend to be persistent and worsen gradually over time. The rate of progression can vary between individuals but eventually leads to respiratory failure if left untreated. Not all individuals experience the same set of symptoms and they may come and go unpredictably.
Risk Factors for IPF
Some significant risk factors that have been identified for developing IPF include:
– Older age (above 50 years): Advancing age is a big risk factor as the lungs lose their elasticity over time.
– Male gender: Men are twice as likely as women to develop Idiopathic Pulmonary Fibrosis.
– Cigarette smoking: Smokers or former smokers are at much higher risk compared to non-smokers.
– Gastroesophageal reflux disease (GERD): Long-standing acid reflux disease damages the lungs over time.
– Environmental exposures: Exposure to mineral or metal dust, wood dust, chemical fumes and gases increases IPF risk.
– Genetics: A small percentage show familial patterns but the condition itself isn’t hereditary.
While risk increases with the above factors, an underlying trigger cannot be pinpointed in many IPF cases. Genetic predisposition along with epigenetic changes are believed to cause abnormal wound healing in lungs leading to fibrosis.
Diagnosis of IPF
Establishing an accurate diagnosis of IPF requires a multidisciplinary discussion between a pulmonologist, radiologist and pathologist. Tests used may include:
– High-resolution chest CT scan: Shows characteristic patterns of lung scarring which is subtle in early stages.
– Pulmonary function tests: Helps assess the degree of lung damage by measuring how well lungs take in and release air.
– Lung biopsy: A surgical lung biopsy may be required to rule out other ILDs if CT and clinical features are not typical of IPF.
– Exclusion of known causes: Other causes of pulmonary fibrosis like connective tissue diseases need to be excluded to confirm IPF.
The 2011 diagnostic criteria published by American Thoracic Society help diagnose IPF based on a combination of clinical, radiological and histopathological features. An accurate diagnosis is crucial for choosing the right treatment approach.
Treatment Options for IPF
Currently there is no cure for IPF but available treatments can help slow progression of disease and improve quality of life by reducing symptoms. Main treatment options include:
– Pulmonary rehabilitation: Focuses on exercises to strengthen respiratory muscles and improve endurance.
– Oxygen therapy: Supplemental oxygen helps those with advanced disease breathe more easily.
– Lung transplantation: Considered for individuals under 75 years of good functional status.
– Antifibrotic drugs: Pirfenidone and nintedanib slow progression of disease by around 50% when administered early.
– Additional medications: May be used to manage symptoms like cough, acid reflux and shortness of breath.
– Supportive care: Good nutrition, vaccinations, stress management aids coping with this chronic condition.
Research into newer anti-fibrotic and cell-based therapies continues but current options can help improve outcomes when used under medical guidance according to individual disease severity. Regular follow up is essential as risk of pulmonary infections is higher in IPF patients.
Prognosis of IPF
The prognosis of IPF depends on multiple factors like age at diagnosis, lung function levels at start of treatment and rate of disease progression. Average survival time without a lung transplant is around 3-5 years post-diagnosis though some may survive longer. Anti-fibrotic drugs if started early can potentially extend survival. Close monitoring of lung function is needed to assess response to treatment over time. Proper care, timely interventions, and good respiratory health can help IPF patients maximize quality of life.
Understanding key aspects of what causes IPF, recognising its signs, early diagnosis and optimising available management can provide the best outcomes for individuals living with this progressive lung condition. While a cure remains elusive, ongoing research aims to develop more targeted therapies to better treat IPF in the future.
*Note:
1. Source: Coherent Market Insights, Public sources, Desk research
2. We have leveraged AI tools to mine information and compile it